Bruton's tyrosine kinase (BTK) is a critical enzyme in the B-cell receptor (BCR) signaling pathway, playing a pivotal role in B-cell development, differentiation, and survival. Due to its central role in B-cell malignancies, BTK has become a key therapeutic target, with inhibitors like ibrutinib and acalabrutinib showing significant clinical efficacy in treating chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and other B-cell disorders. However, the emergence of resistance mutations, such as BTK-T474I, poses a significant challenge to the long-term effectiveness of these therapies.
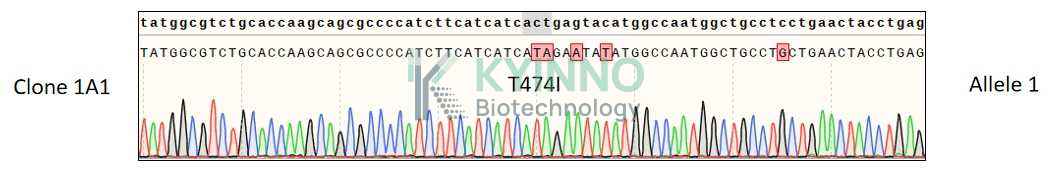
The BTK-T474I mutation is a point mutation in the BTK gene, where threonine at position 474 is replaced by isoleucine. This mutation is located in the kinase domain of BTK, a region critical for the binding of covalent BTK inhibitors. The substitution of threonine with isoleucine alters the steric and electronic properties of the binding pocket, reducing the affinity of covalent inhibitors like ibrutinib. As a result, patients harboring this mutation often exhibit resistance to first-generation BTK inhibitors, leading to disease progression.
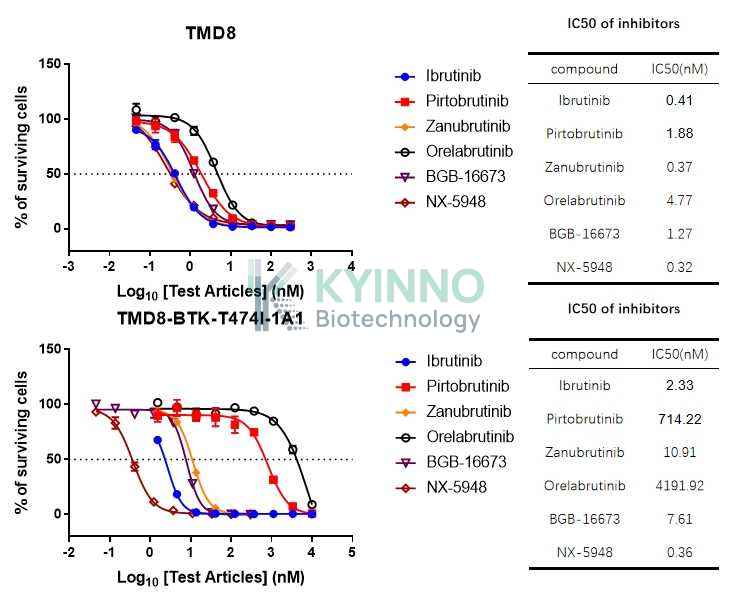
Understanding the structural and functional implications of the BTK-T474I mutation is essential for developing next-generation BTK inhibitors that can overcome resistance. Structural studies have shown that the T474I mutation disrupts the formation of a hydrogen bond between BTK and ibrutinib, which is crucial for the inhibitor's covalent binding. This insight has guided the design of non-covalent BTK inhibitors, such as pirtobrutinib (LOXO-305), which can effectively target both wild-type and mutant forms of BTK, including T474I.
The clinical significance of BTK-T474I underscores the need for ongoing research into resistance mechanisms and the development of novel therapeutic strategies. By elucidating the molecular basis of resistance and designing inhibitors that can circumvent these challenges, researchers aim to improve outcomes for patients with B-cell malignancies.